Blog
Where Do We Stand with the New EU MDR, Additive Manufacturing, and Customised Medical Devices?
One of the growing trends in the medical device industry is customised products for individual patients. An example is an implant designed specifically for an individual patient. A range of technologies make products like this possible, including new additive manufacturing technologies.
Furthermore, customised medical device products offer significant benefits to patients. There are benefits to the MedTech industry too, although customisation also presents challenges.
For medical device manufacturers, product customisation requires a change of strategy, manufacturing processes, and business processes. There are also challenges in terms of regulations.
How do you get regulatory approval for a customised medical device product?
Understanding the Definition of Custom-Made
The starting point is to understand the definition of what EU regulators regard as a custom-made product. Crucially, this definition is changing with the introduction of the new EU Medical Device Regulations (MDR).
Previously, a medical device produced using an additive manufacturing technique (often referred to as 3D printing) and customised for a specific patient would have been considered a custom-made product.
This is no longer the case with the new EU MDR.
A New Definition for Custom-Made Medical Devices in the EU
So, what do the new regulations say in their provision for custom-made medical device products?
In MDR 2017/745, a custom-made product is defined as a medical device that has “specific design characteristics” that makes it suitable “for the sole use of a particular patient exclusively to meet their individual conditions and needs”.
Crucially, the regulations also say that custom-made medical devices must always be produced on the basis of a written prescription provided by a suitably qualified medical professional.
This could cover products made using additive manufacturing techniques and customised for individual patients, but the definition in the regulations doesn’t stop there. They go on to exclude two categories of medical device product from the custom-made definition:
- Mass-produced medical device products which are adapted to the specific requirements of a patient
- Mass-produced products manufactured as per a written prescription
As many customisable medical device products made using additive manufacturing techniques could be defined as mass-produced, they may fall under the above two exclusions. This would mean the new EU MDR does not treat them as being custom-made.
Without further guidance or clarification, the above definition indicates that the label of custom-made, and the resulting regulatory requirements, can only be applied to products made from scratch.
What Does This Mean for the Use of Additive Manufacturing Techniques to Customise Medical Device Products?
In simple terms, more information is needed. Let’s take as an example a mass-produced implant that is 3D printed so that each product is unique. Will it be regarded as custom-made or not?
Under the current definition, it won’t be treated as a custom-made product which might mean it needs its own CE mark and clinical evidence.
In other words, as with many other areas of the new EU MDR, more information is needed.
Solutions Exist
That said, there are solutions when you look at other jurisdictions.
In the US, for example, FDA guidance says customised medical devices can be based on a standard size template for regulatory approval.
Alternatively, the manufacturer can specify a performance or design envelope within which the product will be manufactured. This allows for patient-specific customisation of the medical device within the approved performance/design range.
So, while there are uncertainties now with the new EU MDR, the challenges can be overcome to the benefit of patients as well as to medical device manufacturers looking to expand their product customisation offerings.

8 Essential Elements for De-Risking an Automation Project
Risks can enter an automation project at just about any stage. Those risks can result in serious consequences from budget and schedule overruns to unplanned downtime, disrupted supply chains, and more.
Therefore, it’s important to de-risk your automation project at every possible stage. This ensures errors are reduced to a minimum as well as ensuring the project delivers on expectations.
De-Risking Your Automation Project
At SL Controls, our project delivery processes focus on de-risking at every stage, from the pre-automation phase through to final commissioning and testing.
Central to this are eight essential elements to de-risking all automation projects.
1. Proof of Principle
Proof of Principle (POP) involves proving the feasibility of a proposed solution achieving the desired outcomes for the project. It does this by demonstrating design robustness, performance, and quality characteristics offline so when the design is integrated into the project, confidence of success is higher.
By going through the POP process, we can avoid pursuing goals or targets that are not feasible or achievable and, instead, allow project teams to make changes to achieve the desired outcome. This prevents wasted expenditure.
2. Design for Manufacturing
Design for manufacturing (DFM) is most commonly used when designing new products or major iterations to existing products. It involves ensuring the product’s design makes the product easy to manufacture within cost targets (i.e. targets assigned by leveraging the experience of Subject Matter Experts).
In relation to an automation project, we adapt DFM principles to ensure we don’t just create a fancy automation solution. Instead, our goal is to develop an automation solution that fits user requirements as well as the resources and skillsets available to run and maintain it. It’s also essential the solution we develop matches the budget available, plus we ensure it improves several key metrics, i.e. OEE, safety, quality, etc.
3. User Requirements Definition
A User Requirements Definition is a document we create that specifies what you, as our client, expect the automation project to deliver. It’s not a technical document but should include, among other things:
- Required functions and features of the solution
- Workflow of the solution
- Integration requirements
- Data requirements
- Regulatory requirements
- Life cycle requirements
In terms of the de-risking process, creating and agreeing a User Requirements Definition is part of the project’s feasibility analysis. It also ensures everyone is on the same page.
4. Design Review Management
The design review is an essential part of de-risking automation projects. In regulated sectors, it’s also a compulsory part of the process and must be properly documented.
A design review tests and evaluates the design of the automation solution against the User Requirements Definition. At SL Controls, we insist on peer reviews by multiple Subject Matter Experts to ensure we get a broad spectrum of opinion and can maximise on the experience that exists within the company.
5. Factory Acceptance Testing
The goal of Factory Acceptance Testing (FAT) is to ensure the equipment, platforms, and components of the automation solution deliver on requirements before it leaves the vendor site. It is an essential process that highlights issues and errors, so it plays a key role in de-risking automation projects.
6. Logistics Management
Logistics management is often taken for granted in automation projects, despite the fact there are so many things that can go wrong, many of which can have significant consequences. A good example is a piece of equipment getting dropped, damaged, or delayed. Any of these things could set a project back for weeks or months.
To mitigate these risks, we insist on using qualified and approved freight forwarders who take essential steps to ensure the teardown, crating, and transportation of equipment and machinery goes smoothly. This includes:
- Mapping every aspect of the route
- Planning the equipment required
- Deciding on the crate specification
- Selecting appropriate bracing to secure equipment
- And more
7. Site Acceptance Testing
While the aim of a de-risking strategy is to eliminate risks at the earliest possible stages of an automation project, the Site Acceptance Testing stage is still important. It’s also an essential GMP requirement.
8. Final Commissioning
This includes IQ (installation qualification), OQ (operational qualification), and PQ (performance qualification).
De-Risking Strategy
By emphasizing de-risking in the earliest stages and then focusing on the elimination or mitigation of risks through each subsequent stage, we ensure the successful and smooth delivery of automation projects, maximising the realisation of benefits with respect to safety, yield, OEE, and ROI.

Additive Manufacturing in the Medical Device Industry
Additive manufacturing involves manufacturing a product by fabricating it layer by layer, building it up. It is sometimes referred to as 3D printing, although the technologies involved and the processes that are available are now more advanced than the term 3D printing indicates.
All additive manufacturing techniques differ from traditional manufacturing techniques that produce components/products by removing material (such as in machining) or shaping material (such as extrusion or blow moulding). Instead, additive manufacturing builds up a component or product one tiny layer at a time.
The Benefits of Additive Manufacturing to the Medical Device Industry
- Can produce almost any type of shape, including highly complex and intricate shapes
- Reduces the need to manufacture a product in parts. Instead, additive manufacturing lets you, in many situations, produce the component/product in a single piece, eliminating the need for parts assembly completely.
- The above creates higher quality products
- The point above also makes it possible to create shapes, components, and products that are not possible using other manufacturing techniques
- Can significantly speed up the prototyping process as changes can be implemented and produced quickly
- The speed of making changes also makes additive manufacturing ideal for mass customisation right down to manufacturing batch sizes of one.
Current Applications of Additive Manufacturing in the Medical Device Industry
- Creating models of a patient’s anatomy that surgeons can practice on before real surgery takes place
- To make patient prosthetics
- Manufacture of patient-specific implants
- In dentistry to make dentures and implants
Additive Manufacturing Processes
There is a range of additive manufacturing processes available depending on the component/product you need to manufacture. Those processes include:
- Powder Bed Fusion
- Vat Photopolymerization
- Directed Energy Deposition
- Binder Jetting
- Material Extrusion
- Material Jetting
- Sheet Lamination
Powder Bed Fusion
Powder bed fusion uses melted powder, making it possible to fuse particles together and build up a 3D object layer by layer. In simple terms, material powder is rolled or spread over the previous layer before being fused together with heat.
The main techniques involved are:
- Electron beam melting (EBM)
- Direct metal laser melting (DMLM)
- Directed metal laser sintering (DMLS)
- Selective laser sintering (SLS)
- Selective laser melting (SLM)
- Selective heat sintering (SHS)
Binder Jetting
In the binder jetting additive manufacturing process, powdered material is added to the product in layers. A liquid binder, or adhesive, is applied to each layer. The print head deposits the powdered material and the liquid binder alternately, building up the component or product.
Vat Photopolymerization
In the vat photopolymerization process, a component or product is made from a vat of liquid photopolymer resin. This process can produce components with smooth surfaces and fine details.
It uses a build platform located in a vat of liquid polymer, with the build platform moving down as each new layer is added. In other words, vat photopolymerization works from the top down.
As the build platform moves down each layer, a UV light source cures the photopolymer resin.
Directed Energy Deposition
Directed energy deposition features a nozzle that moves around multiple axes – up to five. This nozzle deposits material onto the component or part in layers. This material can either be in the form of a wire filament or powder. The powder can be metal, ceramic, or polymer.
This material is then fused to the component or part using a laser, plasma arc, or electron beam.
Material Extrusion
Material extrusion is the most common type of 3D printing technology, not least because it is inexpensive and fast.
It involves the material, usually a polymer, passing through a heated nozzle before being added/fused to the component or product as the next layer. The nozzle moves horizontally across the component/product to add the layer, with the build platform moving vertically so the next layer can be added.
Material Jetting
Material jetting works on similar principles to an inkjet printer. A nozzle jets polymer onto the component/product to build up each layer. UV light then cures or hardens the material to finish the layer before moving to the next one.
Sheet Lamination
There are two main sheet lamination processes:
- Ultrasonic additive manufacturing (UAM) – the material is in the form of ribbons or sheets of metal. They are bound together, layer by layer, using ultrasonic welding. The materials that can be used include stainless steel, titanium, aluminium, and copper.
- Laminated object manufacturing (LOM) – this process is similar to the above except the material used is paper. It is bonded layer by layer using adhesive. The result resembles a 3D plywood object.
The Future
Additive manufacturing technologies continue to advance, with potential medical applications in the future that will dramatically enhance patient care and improve treatments. That said, additive manufacturing also offers benefits and opportunities today.

Delivering Bespoke Software Solutions to Companies in the Regulated Sector
As automation, machine learning, sensor technologies, remote monitoring, artificial intelligence, etc advance, manufacturing companies increasingly rely on bespoke software solutions to achieve their objectives. This includes both fully bespoke solutions and software that is customised according to your requirements.
Delivering this type of tailored software to the regulated sector – such as to pharmaceutical or medical device manufacturers – is very different from developing bespoke software for companies in other industries.
Achieving Client Objectives
As with other companies in other industries, software solutions developed for the regulated sector must deliver on client business objectives. This includes:
- Making productivity gains and efficiency savings
- Improving OEE (overall equipment effectiveness)
- Enhancing capacity
- Reducing costs (in relation to maintenance, production, etc)
- Extending capabilities
- And more
However, software solutions developed for the regulated sector must also comply with regulations.
In addition, the regulated sector often also requires software solutions where the primary purpose is compliance with regulations, both new and existing.
A recent example in the pharmaceuticals industry was the introduction of the Falsified Medicines Directive (FMD). Many companies required new software solutions to, for example, adhere to the serialisation requirements of that directive.
Another ongoing example is the introduction of the new EU Medical Device Regulations (MDR). Serialisation of medical device products is a key component of the new MDR, with manufacturers turning to bespoke software as the solution.
Understanding the Regulations
At SL Controls, we have extensive experience developing new, and customising existing, software solutions for pharmaceutical and MedTech industry clients. Therefore, we know the importance of having in-depth in-house knowledge of all regulations that impact our clients’ businesses, particularly those that directly relate to software. This includes 21 CFR Part 11.
This is not enough on its own, however, as the regulatory landscape constantly changes. New regulations are always either out for consultation or approaching their implementation deadline.
As a result, it’s as important to understand broader regulatory trends as it is to understand existing regulations. This helps ensure software solutions are adaptable, flexible, scalable, and future proof.
Beyond the Regulations
While regulations are an important part of your business if you are in a regulated industry like MedTech or pharmaceuticals, they are still only a part. Therefore, successfully developing and implementing software solutions for businesses like yours means being able to do more than write code and understand regulations.
From your point of view, it’s also important you choose a software developer who understands your industry and your business, including the unique pressures, risks, challenges, and limitations you face.
This requires past experience of similar projects. From this experience, software development teams can build an extensive in-house knowledgebase. So, every lesson learned on every project improves the delivery of the next project.
With around 1.5 million engineering hours under our belt at SL Controls, we take a lot of experience and learnings into every project we work on.
Knowledge Sharing and a Learning Culture
In addition, we believe it’s important to have a strong and supported learning culture in the company. This improves skills but it also reduces the risk of information and knowledge becoming hoarded or trapped silos, i.e. where one person learns something on a project that could help future projects, but there is no structure in place for that knowledge to be shared.
At SL Controls, our learning culture, knowledge sharing structures, and past experience mean we don’t start projects for clients in the regulated sector with a blank page, figuring things out as we go. Instead, we hit the ground running, eliminating errors before they are even introduced.
From your perspective, this means your software development and capital investment projects will finish on-time and within budget while also delivering on your objectives.
To find out more about our bespoke software development capabilities at SL Controls, please contact us today.

Qualification of Cloud Infrastructure and SaaS to Demonstrate Regulatory Compliance
Cloud infrastructure and Software as a Service (SaaS) solutions offer significant benefits to manufacturers in the pharmaceutical and medical device industries. Cloud-based solutions, including SaaS, still need to be validated, however, just like your in-house IT infrastructure.
The main difference, of course, is the introduction of third parties to not only provide a service, but to also ensure they manage that service properly. You can’t do this simply based on trust as you are subject to regulations, not the cloud infrastructure or SaaS vendor.
Therefore, you need a clear qualification strategy to ensure you can benefit from cloud computing and SaaS technologies while also ensuring regulatory compliance.
The Same Objectives
When qualifying cloud infrastructure and SaaS solutions, the objectives are the same as when qualifying in-house IT infrastructure or software that runs on your own system. Those objectives include:
- Cloud and SaaS solutions must be specified;
- They must be qualified to demonstrate they work as intended;
- They must be subject to change control;
- Written procedures must be created;
- Records must be kept; and,
- Staff must be trained.
As you are responsible for ensuring all your systems are compliant, including cloud computing and SaaS elements, the principles of qualification are the same. That said, innovative solutions are often required.
For example, one of the advantages of both cloud computing and SaaS is that vendors constantly work to improve their products, releasing frequent updates. Unlike in-house infrastructure, you are not in control of the application of these updates.
An automated testing process might be the solution in the above example. After all, regulators do not stipulate manual testing completed by humans. Instead, regulations require you to provide evidence the solution performs as expected. A software programme can perform this task in relation to cloud infrastructure or SaaS, automating the process and making it significantly less time-consuming.
Effective Qualification Strategy for Cloud Infrastructure and SaaS Solutions
Whatever the specifics of qualifying cloud infrastructure and SaaS solutions, you need a clear strategy in place first. This strategy should include:
- Vendor assessments – this is required from a technical point of view to ensure the vendor’s solution can deliver on the requirements of the project. However, you should also assess the vendor’s quality system to confirm and demonstrate they have proper procedures in place for things like system security, data integrity, data recovery, change management, staff training, and more. If the vendor has already qualified their solution, the process should be easier as you should only have to check the qualification documentation.
- Risk assessments – you should take a risk-based approach to all stages of qualifying cloud or SaaS solutions. This includes completing risk assessments on things like security and data integrity.
- Qualification plan – this should outline the approach to qualification and detail activities, tasks, and responsibilities.
Taking Advantage
Broadly speaking, the pharmaceutical and medical device industries are behind other industries in their adoption of cloud computing technologies or the SaaS delivery models. It’s understandable that solutions like these have been viewed in the past as adding an element of risk to manufacturing operations.
Today, however, solutions and practices exist that make it possible to actually reduce risk by switching to cloud infrastructure and SaaS solutions. This includes qualification solutions that demonstrate regulatory compliance equal to in-house systems.

Continuous Manufacturing vs Batch Manufacturing in the Pharmaceutical Industry
Batch manufacturing is the tried, tested, and trusted approach for manufacturing pharmaceutical products. There is an increasing push, however, for the industry to switch to continuous manufacturing.
A key driver of this push is the FDA – the Food and Drug Administration in the US. The FDA believes continuous manufacturing improves product quality, reduces medicine shortages, and enhances efficiencies, among other things.
So, should you modernise by moving to continuous manufacturing processes, or should you stay with batch manufacturing?
Batch Manufacturing Explained
Batch manufacturing involves manufacturing pharmaceutical products in multiple steps. At the end of each step, production stops before the process moves to the next step. Hold times like this can vary depending on the next stage of the process.
For example, in some situations, materials are shipped to a different location between steps. For some pharmaceutical products, this results in production times being measured in weeks or months.
Advantages of Batch Manufacturing
The main benefit of batch manufacturing is the fact it is well established, and it works. In addition, pharmaceutical companies have approval from regulators for their products based on them being produced using batch manufacturing techniques.
You probably also have an established and effective supply chain linked to your batch manufacturing process.
For all these reasons, changing from batch manufacturing to continuous manufacturing for existing processes is a significant undertaking. This is before you even take into account the investment required in new equipment, the risks of introducing a new process, and the time it will take to develop the new process, train your team, etc.
Finally, there is also the fact that batch manufacturing is the only viable method to produce certain drugs, although this may change as new technologies are developed.
Disadvantages of Batch Manufacturing
While there are benefits to batch manufacturing, there are downsides too. The length of time it takes to produce a product using batch manufacturing is one of those downsides. Hold times also increase the risk of material degradation, while scaling production can be a challenge.
The latter particularly applies to scaling up production. To scale up production in a batch manufacturing process, you typically need to scale up your equipment. This requires time, money, and additional space.
Continuous Manufacturing Explained
The pharmaceutical industry stands out from many other manufacturing industries that have been using the continuous manufacturing technique for decades.
Continuous manufacturing involves a pharmaceutical product being produced nonstop in one continuous manufacturing process. The entire process takes place in one facility, start to finish, without hold times.
Advantages of Continuous Manufacturing
The benefits of continuous manufacturing include:
- Reduced manufacturing costs, particularly over the long term
- Shorter production times – reducing manufacturing times from weeks to days is not unusual
- Reduces the risk of human error
- Improves quality
- Monitoring is more efficient as continuous manufacturing processes typically use automated monitoring techniques and predictive maintenance
In addition, continuous manufacturing is a more agile technique, making scaling production much easier. Scaling up, for example, simply means running the continuous manufacturing process for longer.
In other words, matching supply to demand is much easier with continuous manufacturing than it is with batch manufacturing
Continuous manufacturing also makes track and trace more flexible, i.e. instead of a batch being defined by the equipment used in its manufacture, you can instead define a drug’s batch quantity based on things like a period of time or the quantity manufactured. This makes it possible to have smaller batch sizes which means less waste when defects are detected in a batch.
Is Continuous Manufacturing the Future for the Pharmaceutical Industry?
While the move towards the use of continuous manufacturing techniques has been slow, more and more facilities are choosing continuous manufacturing processes when introducing a new product.
In addition, regulators continue to allay fears in the industry regarding approval delays for drugs produced using continuous manufacturing techniques.
Plus, the benefits of continuous manufacturing are too hard to ignore.
At least for new products and new facilities, continuous manufacturing is increasingly the future for the pharmaceutical industry.

The UDI and Serialisation Requirements of the New EU MDR
The clock is well as truly ticking on the introduction of the new EU MDR (Medical Device Regulations). The process of implementing this new EU directive started in 2017, with it scheduled to come into force on 26 May 2020, i.e. less than a year from now.
EU MDR represents a significant change to regulations governing the development, regulatory approval, and manufacture of medical device products. It has several different aspects including:
- Many products are being reclassified using a new risk-based classification system
- There are now stricter pre-market controls
- There is a new requirement for medical device product manufacturers to establish a post-market surveillance system for the products they produce
- And more
One of the key areas that is changing under the new EU MDR, however, is the requirement to have Unique Device Identifiers (UDI) on all medical devices.
Unique Device Identifiers
The concept of including UDIs on the labels of medical device products is not new, of course. After all, it has been necessary to include UDIs on MedTech products marketed in the US since 2013.
The new European regulations are more stringent, however, as they will apply to a broader range of products than the rules in the US.
If you already ship to the US or manufacture according to FDA standards on UDIs, you will have less to do to achieve compliance with the new EU MDR. However, you will still have to make changes.
In other words, the new UDI rules will impact all medical device manufacturers making products for the EU market. Here are the key things you need to know.
Why is There a UDI Element in the New EU MDR?
There are three main reasons for including a UDI element in the new EU MDR:
- Improves track and trace capabilities through the entire product lifecycle
- Combats counterfeiting
- Enhances transparency
What Are the New UDI Requirements?
In very brief terms, the new UDI requirements involve creating a UDI for each medical device product you manufacture. You must send this UDI to the EU Database for Medical Devices (EUDAMED).
In addition, the UDI must also be part of the product label of each medical device.
When Do the New Rules Apply?
- Sending UDI information to the EUDAMED – you must do this by May 2020 when the rest of the new EU MDR comes into force
- UDI included on product labels for Class III products and implants – more time is being given to manufacturers to include UDIs on product labels given the scale of the challenge this can present. For Class III products, the deadline is May 2021.
- UDI included on product labels for Class IIa and IIb products – May 2023
- UDI included on product labels for Class I products – May 2025
What Are the UDI Requirements?
- The UDI must appear in plain text on all packaging parts of a product;
- It must be machine-readable; and,
- It must feature two components – a Device Identifier (DI) and a Production Identifier (PI or GTIN).
What Does the UDI-DI Element Contain?
The UDI-DI identifies the product, plus it contains the product’s master data including:
- Product name
- Manufacturer name
- Type of control
- Quantity in a pack
- Size
- Warning notices (next to the serialisation requirements, this is one of the biggest impacts of the new UDI rules)
What Does the Production Identifier Contain?
- Date of manufacture
- Expiry date
- Serial number or lot number
- And more
The serial number for some products will need to be an individual serial number. For others, however, the serial number will be a batch number. Either way, this represents a substantial change to the previous system.
The Serialisation Solution to the UDI Requirements of EU MDR
For most medical device manufacturers, the most effective way to comply with the new UDI requirements of EU MDR is to implement a new serialisation solution or update an existing one.
This is necessary because of the large amount of new information that is required on medical device product labels. It’s also necessary because the new requirements present a significant data collection, storage, and transmission challenge for MedTech manufacturers.
Working with a specialist serialisation solution provider will ensure a smooth transition to compliance with the new regulations. Find out more by contacting SL Controls today.
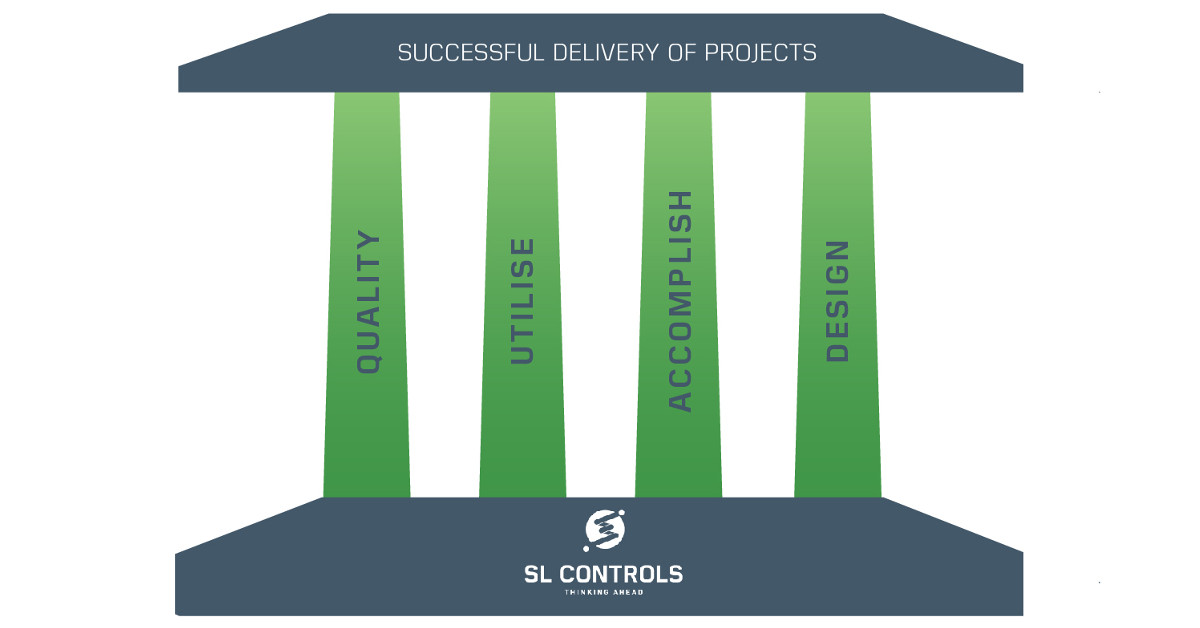
Infographic – The Four Pillars of Successful Project Delivery
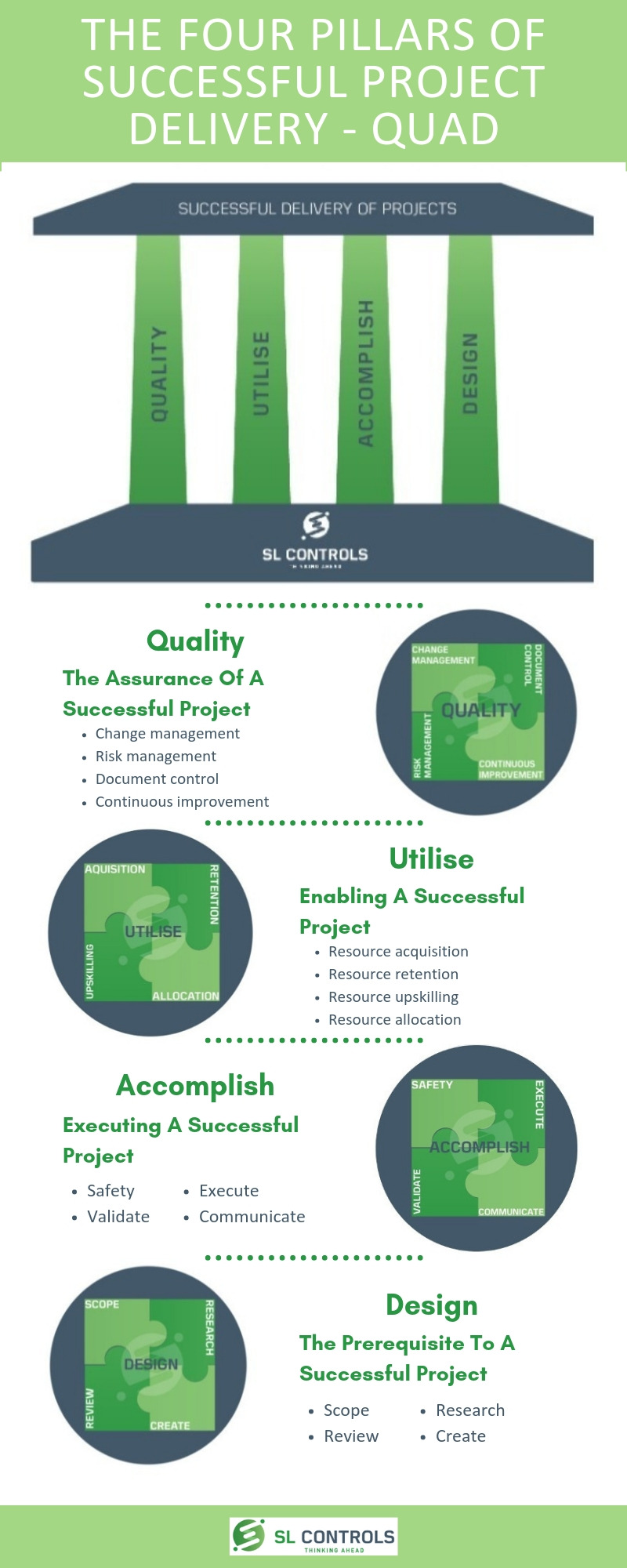
At SL Controls, we describe the four pillars of successful project delivery as QUAD:
- Q – Quality
- U – Utilise
- A – Accomplish
- D – Design
Quality – The Assurance of a Successful Project
- Change management
- Risk management
- Document control
- Continuous
Utilise – Enabling a Successful Project
- Resource acquisition
- Resource retention
- Resource upskilling
- Resource allocation
Accomplish – Executing a Successful Project
- Safety
- Validate
- Execute
- Communicate
Design – The Prerequisite to a Successful Project
- Scope
- Review
- Research
- Create
How SL Controls’ Investment in Ireland’s Regions Benefits MedTech and Pharmaceutical Manufacturers
SL Controls is a company that has deep roots in Sligo. This is where the company was first founded, it’s where we employed our first staff, and it’s the location of our head office.
While Sligo remains crucial to our business operations, we now also have other locations around Ireland that are of equal importance. Our Irish offices now include:
- Sligo
- Limerick
- Galway
- Maynooth/Dublin
If you are in either the MedTech or pharmaceutical sectors in Ireland, you will recognise each of these locations as being key regional hubs.
In Sligo, Limerick, Galway, and Maynooth/Dublin, there are both indigenous Irish and internationally-based manufacturers of pharmaceuticals and/or medical devices. In addition, each of the locations also has a strong ecosystem of suppliers, companies providing a range of support services, and educational institutions.
Ireland isn’t a large country, though. Plus, new technologies increasingly enable efficient remote collaborative working without the need to travel at all. Why, then, did SL Controls decide to invest so heavily in Ireland’s regions?
Investing in MedTech and Pharmaceutical Regional Hubs
We identified two major benefits for investing in the main MedTech and pharmaceutical regional hubs in Ireland:
- Benefits to our team, many of which also indirectly benefit our clients
- Direct benefits to our clients
We explored other options too, of course, including increasing the numbers of people who work remotely. The above benefits were too compelling to ignore, however.
Plus, it’s working extremely well now the regional offices are open and established.
Benefits to Our Team
Improving work-life balance is a key priority for most of the Irish workforce. One crucial aspect of achieving the right balance is to bring business travel, daily commutes, and nights spent away from home to a minimum.
Before we opened our regional offices, some of our team had to commute longer distances than they wanted to. In addition, to fulfil client contracts, our engineers had to spend multiple days in the field, staying in hotels and spending significant hours driving.
Enabling members of our team to live and work closer to our key clients has been an important driver of the recent success we have had at SL Controls.
This is a better model than large-scale remote working arrangements too, as our people feel more of a connection to the company as a whole, plus there is a strong sense of team in each of our locations.
As a result, individuals get the support they need, our projects run more efficiently, and it’s easier to share knowledge – something which is crucial in our industry.
Benefits to Industry
Our clients also benefit directly from the fact we have offices in key MedTech and pharmaceutical regional hubs in Ireland. Those benefits include:
- Improved relationships with key members of our team
- Excellent staff retention rates at SL Controls which provides consistency and familiarity
- Projects run more efficiently as teams spend more time working together and less time travelling
- We can respond faster to client requests
- It’s easier to get essential expertise on-site when you need it
The Future
Our decision to invest in Ireland’s regions benefits SL Controls, our team, and our clients. We also strive to ensure it benefits the local communities where our offices are located too. We actively take part in the business community in our regional locations, we support local causes, and provide other support any way we can.
Our future at SL Controls is in Ireland’s regions, so our investment in those regions continues.
« Previous 1 … 10 11 12 13 14 … 18 Next »


